 891
891 THERMAL ANALYSIS
THERMAL ANALYSIS
Precisely determined thermodynamic events, such as a change of state, can indicate the identity and purity of drugs. Compendial standards have long been established for the melting or boiling temperatures of substances. These transitions occur at characteristic temperatures, and the compendial standards therefore contribute to the identification of the substances. Because impurities affect these changes in predictable ways, the same compendial standards contribute to the control of the purity of the substances.
Thermal analysis in the broadest sense is the measurement of physical-chemical properties of materials as a function of temperature. Instrumental methods have largely supplanted older methods dependent on visual inspection and on measurements under fixed or arbitrary conditions, because they are objective, they provide more information, they afford permanent records, and they are generally more sensitive, more precise, and more accurate. Furthermore, they may provide information on crystal perfection, polymorphism, melting temperature, sublimation, glass transitions, dehydration, evaporation, pyrolysis, solid-solid interactions, and purity. Such data are useful in the characterization of substances with respect to compatibility, stability, packaging, and quality control. The measurements used most often in thermal analysis, i.e., transition temperature, thermogravimetry, and impurity analysis, are described here.
Transition Temperature—
As a specimen is heated, its uptake (or evolution) of heat can be measured [differential scanning calorimetry (DSC)] or the resulting difference in temperature from that of an inert reference heated identically [differential thermal analysis (DTA)] can be measured. Either technique provides a record of the temperature at which phase changes, glass transitions, or chemical reactions occur. In the case of melting, both an “onset” and a “peak” temperature can be determined objectively and reproducibly, often to within a few tenths of a degree. While these temperatures are useful for characterizing substances, and the difference between the two temperatures is indicative of purity, the values cannot be correlated with subjective, visual “melting-range” values or with constants such as the triple point of the pure material.
A complete description of the conditions employed should accompany each thermogram, including make and model of instrument; record of last calibration; specimen size and identification (including previous thermal history); container; identity, flow rate, and pressure of gaseous atmosphere; direction and rate of temperature change; and instrument and recorder sensitivity.
It is appropriate to make a preliminary examination over a wide range of temperature (typically room temperature to decomposition temperature or about 10
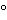
to 20
above the melting point) and over a wide range of heating rates (2
to 20
per minute), which may reveal unexpected effects; then a single examination or replicate examinations over a narrow range, bracketing the transition of interest at one or more lower heating rates, can be made. In examining pure crystalline materials, rates as low as 1
per minute may be appropriate, whereas rates of up to 10
per minute are more appropriate for polymeric and other semi-crystalline materials. As the reliability of the measurements varies from one substance to another, statements of the number of significant figures to be used in the reporting of intralaboratory repeatability and of interlaboratory reproducibility cannot be given here, but should be included in the individual monograph.
Thermogravimetric Analysis—
Thermogravimetric analysis involves the determination of the mass of a specimen as a function of temperature, or time of heating, or both, and when properly applied, provides more useful information than does loss on drying at fixed temperature, often for a fixed time and in what is usually an ill-defined atmosphere. Usually, loss of surface-absorbed solvent can be distinguished from solvent in the crystal lattice and from degradation losses. The measurements can be carried out in atmospheres having controlled humidity and oxygen concentration to reveal interactions with the drug substance, between drug substances, and between active substances and excipients or packaging materials.
While the details depend on the manufacturer, the essential features of the equipment are a recording balance and a programmable heat source. Equipment differs in the ability to handle specimens of various sizes, the means of sensing specimen temperature, and the range of atmosphere control. Calibration is required with all systems, i.e., the mass scale is calibrated by the use of standard weights; calibration of the temperature scale, which is more difficult, involving either variations in positioning of thermocouples and their calibration; or in other systems, calibration involves the use of standard materials because it is assumed that the specimen temperature is the furnace temperature.
Procedural details are specified in order to provide for valid interlaboratory comparison of results. The specimen weight, source, and thermal history are noted. The equipment description covers dimensions and geometry, the materials of the test specimen holder, and the location of the temperature transducer. Alternatively, the make and model number of commercial equipment are specified. In all cases, the calibration record is specified. Data on the temperature environment include the initial and final temperatures and the rate of change or other details if nonlinear. The test atmosphere is critical; the volume, pressure, composition, whether static or dynamic, and if the latter, the flow rate and temperature are specified.
Eutectic Impurity Analysis—
The basis of any calorimetric purity method is the relationship between the melting and freezing point depression, and the level of impurity. The melting of a compound is characterized by the absorption of latent heat of fusion,
DHf, at a specific temperature,
To. In theory, a melting transition for an absolutely pure crystalline compound should occur within an infinitely narrow range. A broadening of the melting range, due to impurities, provides a sensitive criterion of purity. The effect is apparent visually by examination of thermograms of specimens differing by a few tenths percent in impurity content. A material that is 99% pure is about 20% molten at 3
below the melting point of the pure material (see
accompanying figure).
Superimposed Thermograms Illustrating the Effect of Impurities on DSC Melting Peak Shape
The parameters of melting (melting range, DHf, and calculated eutectic purity) are readily obtained from the thermogram of a single melting event using a small test specimen, and the method does not require multiple, precise actual temperature measurements. Thermogram units are directly convertible to heat transfer, millicalories per second.
The lowering of the freezing point in dilute solutions by molecules of nearly equal size is expressed by a modified van't Hoff equation:
in which
T = absolute temperature in degrees Kelvin (
K),
X2 = mole fraction of minor component (solute; impurity),
DHf = molar heat of fusion of the major component,
R = gas constant, and
K = distribution ratio of solute between the solid and liquid phases.
Assuming that the temperature range is small and that no solid solutions are formed (K = 0), integration of the van't Hoff equation yields the following relationship between mole fraction of impurity and the melting-point depression:
in which
To = melting point of the pure compound, in
K, and
Tm = melting point of the test specimen, in
K.
With no solid solution formation, the concentration of impurity in the liquid phase at any temperature during the melting is inversely proportional to the fraction melted at that temperature, and the melting-point depression is directly proportional to the mole fraction of impurity. A plot of the observed test specimen temperature,
Ts, versus the reciprocal of the fraction melted, 1/
F, at temperature
Ts, should yield a straight line with the slope equal to the melting-point depression (
To 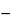 Tm
Tm). The theoretical melting point of the pure compound is obtained by extrapolation to 1/
F = 0:
Substituting the experimentally obtained values for
To Tm,
DHf, and
To in
Equation 2 yields the mole fraction of the total eutectic impurity, which, when multiplied by 100, gives the mole percentage of total eutectic impurities.
Deviations from the theoretical linear plot also may be due to solid solution formation (
K 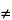
0), so that care must be taken in interpreting the data.
To observe the linear effect of the impurity concentration on the melting-point depression, the impurity must be soluble in the liquid phase or melt of the compound, but insoluble in the solid phase, i.e., no solid solutions are formed. Some chemical similarities are necessary for solubility in the melt. For example, the presence of ionic compounds in neutral organic compounds and the occurrence of thermal decomposition may not be reflected in purity estimates. The extent of these theoretical limitations has been only partially explored.
Impurities present from the synthetic route often are similar to the end product, hence there usually is no problem of solubility in the melt. Impurities consisting of molecules of the same shape, size, and character as those of the major component can fit into the matrix of the major component without disruption of the lattice, forming solid solutions or inclusions; such impurities are not detectable by DSC. Purity estimates are too high in such cases. This is more common with less-ordered crystals as indicated by low heats of fusion.
Impurity levels calculated from thermograms are reproducible and probably reliable within 0.1% for ideal compounds. Melting-point determinations by scanning calorimetry have a reproducibility with a standard deviation of about 0.2
. Calibration against standards may allow about 1
accuracy for the melting point, so that this technique is comparable to other procedures.
Compounds that exist in polymorphic form cannot be used in purity determination unless the compound is completely converted to one form. On the other hand, DSC and DTA are inherently useful for detecting, and therefore monitoring, polymorphism.
Procedure—
The actual procedure and the calculations to be employed are dependent on the particular instrument used. Consult the manufacturer's literature and/or the thermal analysis literature for the most appropriate technique for a given instrument. In any event, it is imperative to keep in mind the limitations of solid solution formation, insolubility in the melt, polymorphism, and decomposition during the analysis.
;)
;)
;)
;)