Procedure—
Melt the sample, if it is not already liquid.
[note—The temperature during melting should not exceed the melting point of the sample by more than 10
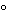
.
] Pass through two pieces of filter paper to remove any solid impurities and the last traces of moisture. The filtration may be performed in an air oven at 100
but should be completed within 5 minutes ± 30 seconds. The sample must be absolutely dry. All glassware must be absolutely clean and completely dry. After filtration, allow the filtered sample to achieve a temperature of 68
to 71 ± 1
before weighing the sample. Once the sample has achieved a temperature of 68
to 71 ± 1
, immediately weigh the sample into a 500-mL iodine flask, using the weights and weighing accuracy noted in the accompanying table.
[note—The weight of the substance must be such that there will be an excess of
iodochloride TS of 50% to 60% of the amount added, that is, 100% to 150% of the amount absorbed.
] Add 15 mL of a fresh mixture of cyclohexane and glacial acetic acid (1:1), and swirl to dissolve the sample. Add 25.0 mL of iodochloride TS, insert the stopper securely in the flask, and swirl to mix. Allow it to stand at 25 ± 5
, protected from light, with occasional shaking, for 1.0 or 2.0 hours, depending on the Iodine Value (IV) of the sample: IV less than 150, 1.0 hour; IV equal to or greater than 150, 2.0 hours. Then, within 3 minutes after the indicated reaction time, add, in the order named, 20 mL of
Potassium Iodide Solution and 150 mL of recently boiled and cooled water, and mix. Within 30 minutes, titrate the liberated iodine with 0.1 N sodium thiosulfate VS, while stirring by mechanical means after each addition of thiosulfate. When the yellow iodine color has almost disappeared, add 1 to 2 mL of
Starch Indicator Solution, and continue the titration with 0.1 N sodium thiosulfate VS until the blue color is discharged. Perform a blank test at the same time with the same quantities of the same reagents and in the same manner (see
Residual Titrations  541
541
). The difference between the volumes, in mL, of 0.1 N sodium thiosulfate consumed by the blank test and the actual test, multiplied by 1.269 and divided by the weight, in g, of the sample taken, is the Iodine Value.
;)
;)